


Modeling of nano-structures / 1st Workshop on Characterization and Analysis of Nanomaterials University of Aveiro, Portugal, July 9, 2019

Prof. Dr. José Coutinho (Workshop coordinator )
jose.coutinho@ua.pt
Prof. Dr. Vladimir Bystrov (Workshop coordinator)
vsbys@mail.ru
Recent developments of computational methods allow us to predict the structure, stability, and properties of a wide range of materials, including as-yet-unreported ones. Promising materials have been explored via high-throughput screening within either publicly available computational databases or unexplored composition and structure space. Reported examples include the identification of nitride semiconductors, TCOs, solar cell photo-absorber materials, and photocatalysts, some of which have been experimentally verified. Machine learning in combination with first-principles calculations has emerged as a technique to accelerate and enhance new discoveries. A blend of computation and experimentation with data science toward the development of materials is often referred to as materials informatics and is currently attracting growing interest.
Calculation “engines” based on standard local and semi-local approximations to density functional theory (DFT) have played an important role in understanding the electronic, optical and magnetic properties of materials. Unfortunately, local-DFT often does not provide sufficient accuracy, unraveling the need to use higher-level methods like the non-local functionals, the random-phase approximation or many-body perturbation methods.
 Figure 1. Atomistic model used to investigate the electron transfer between a material based on Si-nanocrystals and molecular dopants (F4-TCNQ). See “Resonant electronic coupling enabled by small molecules in nanocrystal solids”, R. N. Pereira et al., Nano Letters 14, 3817 (2014); doi:10.1021/nl500932q
Figure 1. Atomistic model used to investigate the electron transfer between a material based on Si-nanocrystals and molecular dopants (F4-TCNQ). See “Resonant electronic coupling enabled by small molecules in nanocrystal solids”, R. N. Pereira et al., Nano Letters 14, 3817 (2014); doi:10.1021/nl500932q
 Figure 2. Real part (a) and imaginary part (b) of the frequency-dependent dielectric function of hydroxyapatite, obtained using local-DFT, non-local-DFT and many-body perturbation theory within the GW approximation.
Figure 2. Real part (a) and imaginary part (b) of the frequency-dependent dielectric function of hydroxyapatite, obtained using local-DFT, non-local-DFT and many-body perturbation theory within the GW approximation.
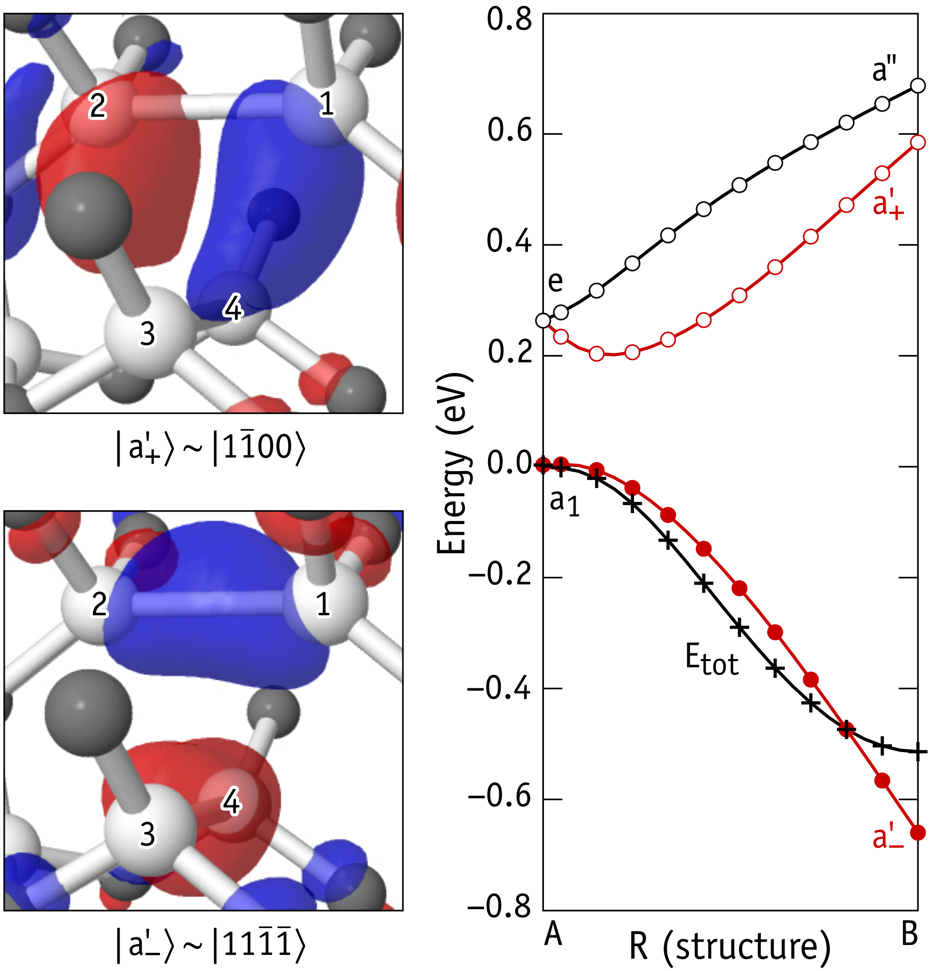 Figure 3. First-principles calculation of the highest occupied and lowest unoccupied states (a’- and a’+) of the carbon vacancy in 4H-SiC (left). On the right, the origin of the pseudo-Jahn-Teller effect is illustrated as an increase of the resonance between a’- and a’+ states as the local distortion increases.
Figure 3. First-principles calculation of the highest occupied and lowest unoccupied states (a’- and a’+) of the carbon vacancy in 4H-SiC (left). On the right, the origin of the pseudo-Jahn-Teller effect is illustrated as an increase of the resonance between a’- and a’+ states as the local distortion increases.